Research
Our research on the specialized plant metabolism combines molecular biology and computational methods to address questions around the evolution and regulation of biosynthesis pathways.
Specialized Metabolism
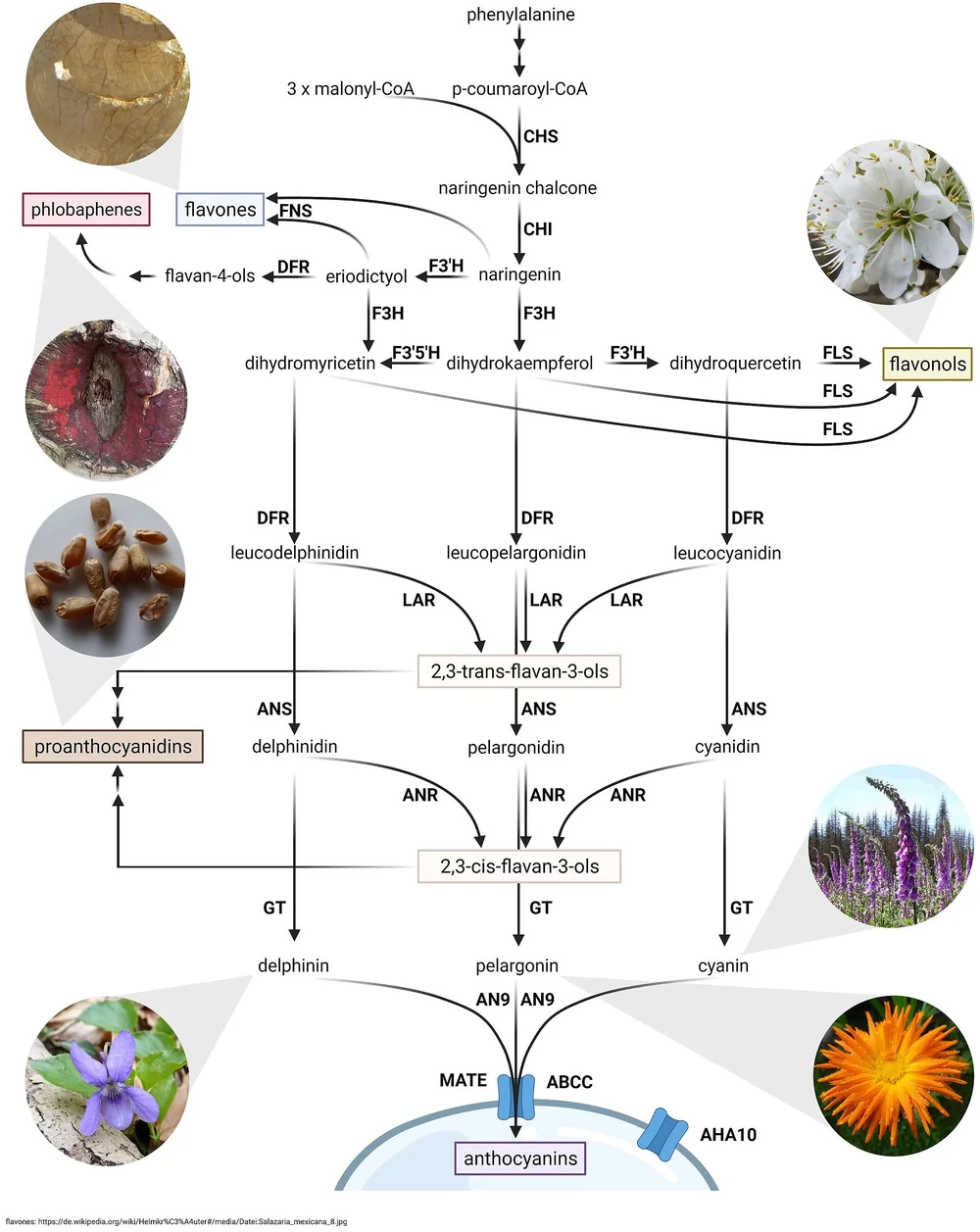
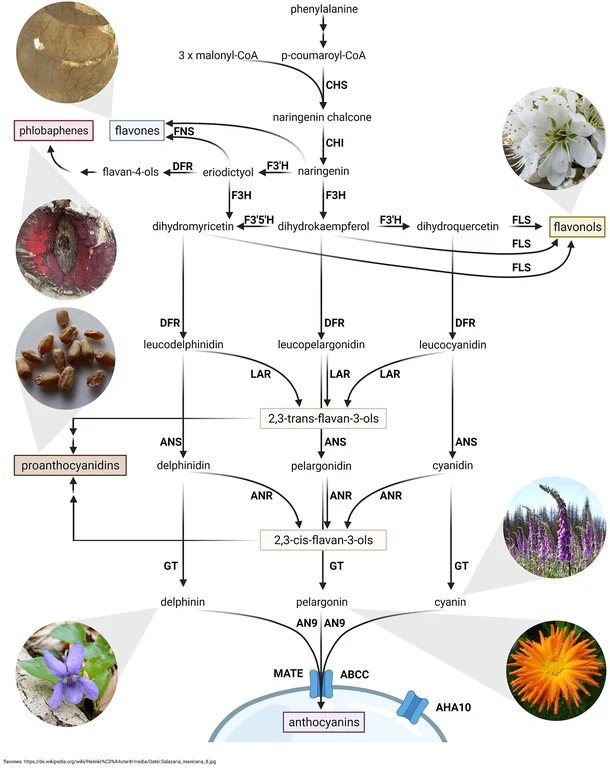
Plants produce a plethora of specialized metabolites to cope with numerous environmental challenges. Examples are terpenoids, flavonoids, and betalains. We are particularly interested in flavonoids which are responsible for the pigmentation of many blue or red flowers. Flavonoids can be separated into several subgroups including flavonols, anthocyanins, proanthocyanidins, and flavones. There are multiple reasons why working on the flavonoid biosynthesis is promising. [Details about flavonoid biosynthesis]
Generally, we are interested in the discovery of promising biosynthesis pathways for biotechnological applications. This is not restricted to the flavonoid biosynthesis, but instead using this model system to develop new genome mining approaches. Different methods for the identification biosynthetic pathways are combined including screens and comparisons of plant genome sequences. Currently, we explore the withanolide biosynthesis in collaboration with the Franke lab (Leibniz University Hannover). [Details about withanolide biosynthesis]
Biosynthetic networks are controlled by transcription factors. One of the largest transcription factor families in plants is the MYBs, which are involved in controlling a variety of plant-specific processes. [Details about MYBs]
Plant Genomics & Long Read Sequencing
Plant genome sequences contain the blue print for all proteins (enzymes). Sequencing and investigating genomes is an effective approach to reveal the biochemical potential of plants. Especially the correlation of genomic data (DNA) with transcriptomic (RNA) and metabolomic (chemical compounds) data sets allows the identification of biosynthesis pathways. Rapid developments of long read sequencing technologies allow the cost-effective analysis of large plant genomes. Sequencers distributed by Oxford Nanopore Technologies (ONT) are portable and can even be operated in the field. This so called nanopore sequencing approach analysis individual DNA strands. We use this technology to resolve the genome sequences of of important plant species. This is also a great opportunity for students to contribute to a genome sequencing project. [Details about plant genome sequencing or our plant genome sequencing project]
Applied Bioinformatics
Specific biological questions require the development of dedicated tools. We write such tools mostly in Python and R. The developed tools are freely available on github (bpucker). Some tools are available on our web server. The following tools are examples of active developments.
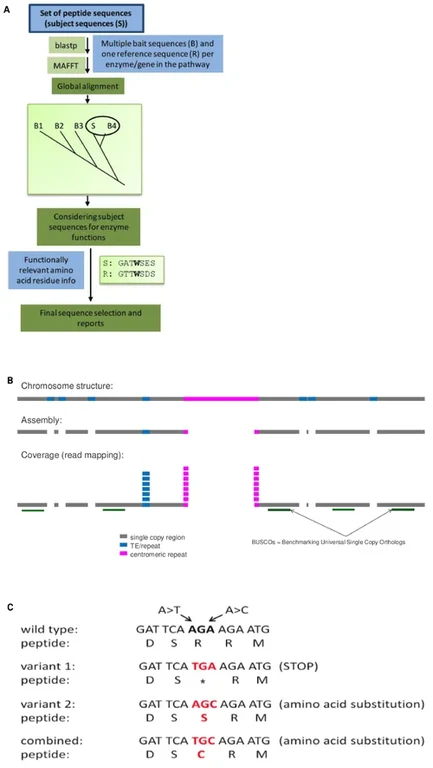
- KIPEs (A): Knowledge-based Identification of Pathway Enzymes allows the automatic annotation of the proteins involved in the core steps of the flavonoid biosynthesis. This supports the identification of molecular mechanisms underlying color differences between cultivars of the same species. In addition, rapid annotation of flavonoid biosynthesis genes in novel/uncharacterized species becomes convenient.
- MGSE (B): Mapping-based Genome Size Estimation is a novel approach to infer the true genome size of a species based on sequence reads. This approach harnesses the equal representation of all regions (even repeats) in the read set. The average number of sequence reads (coverage) is estimated based on single copy regions in a reference genome sequence. Dividing the combined coverage of all positions in an assembly by this sequencing depth results in the genome size estimation.
- NAVIP (C): Neighborhood-Aware Variant Impact Predictor enables the prediction of functional consequences arising from sequence variants between a sequenced sample and a reference. In contrast to many established tools, NAVIP considers all variants in one gene at the same time when predicting the potential effect of sequence variants.
- MYB_annotator (D): This tool enables the automatic identification and annotation of MYBs in a novel transcriptome/genome sequence assembly of a plant species. The identified candidates are functionally annotated based on orthology to previously characterized sequences.
- bHLH_annotator (E): This tool enables the automatic identification and annotation of bHLHs in a novel transcriptome/genome sequence assembly of a plant species. The identified candidates are functionally annotated based on orthology to previously characterized sequences.
- CoExpPhylo (F): This tool eluciates biosynthesis pathways by integrating coexpression data with phylogenetic signals.
- DupyliCate (G): This tool identifies, classifies, and characterizes gene duplications.
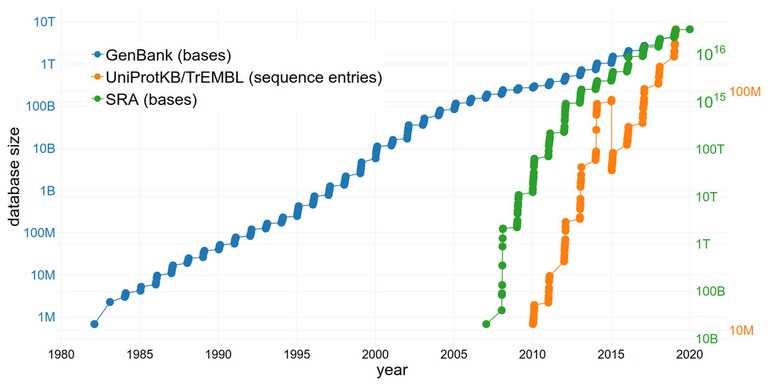
BigData & Databolomics
We integrate public datasets, often sequencing data, into our research projects and utilize them in innovative ways (DataReuse/DataUpcycling). This allows us to address questions not just for a single plant, but directly across all species sequenced to date. However, a common problem is the lack of metadata, meaning information about the origin of samples and the process of data generation. Therefore, we are developing methods to infer missing metadata from the data itself.
A significant challenge in the medium term will be elucidating gene functions. While technological advances in sequencing have made genome decoding easier, functional annotation of individual genes remains a labor-intensive process. Because it is not always possible to elucidate gene functions by knocking out the gene in question, we are developing methods for automatic transfer of information between different plant species. The long-term goal is to integrate all available knowledge on gene functions in plants. Our particular focus is on genes that are active in specialized metabolism.
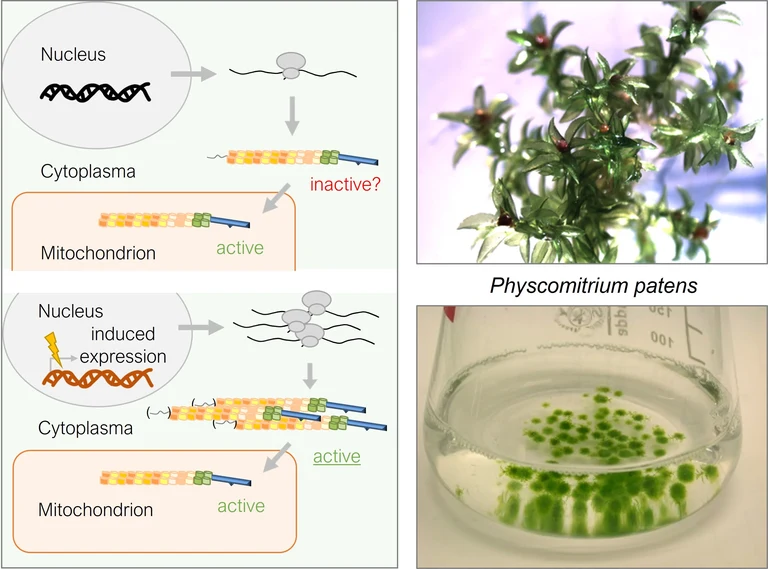
The model moss Physcomitrium patens as expression system for modified and heterologous (RNA-binding) proteins
Physcomitrium patens is a moss with a completely sequenced and well annotated genome and transcriptome. Unlike most other plants, the moss can easily be genetically modified using homologous recombination. This allows stable and precise integration of genetic constructs into the nuclear genome or the modification of single or multiple genomic loci (Rensing et al., 2020). The stable transformation of Physcomitrium patens to introduce promoter construct cassettes (Oldenkott et al., 2020, Lesch et al., 2024) is well established in the lab. In land plants, mitochondrial and chloroplast genomes have different numbers of thymidine (T)-to-cytidine (C) mutations that are corrected at RNA level by C-to-U editing. The different editing sites are targeted by specific pentatricopeptide repeat proteins consisting of a transit peptide for organellar import, a PPR RNA-binding domain and a C-terminal deaminase domain (reviewed in Small et al., 2020). P. patens has been used extensively to investigate C-to-U RNA editing properties of different pentatricopeptide repeat (PPR) proteins. Editing factors from evolutionary distant flowering plants like Macadamia integrifolia or Arabidopsis thaliana have been successfully introduced into the genetic background of P. patens to test their editing capacity (Oldenkott et al., 2020). A just recently established inducible expression system is even better suited to study the function of modified PPR proteins, protein chimeras and synthetic factors in the cytosol of P. patens (Thielen et al., 2024). This line of research is carried out by Dr. Mareike Schallenberg-Rüdinger, mainly as part of Bachelor courses.

Molecular Biology Methods
- High molecular weight DNA extraction (doi: 10.17504/protocols.io.bcvyiw7w)
- Numerous PCR types (long range, colony, nested, cross-over)
- Recombinant DNA technology ('cloning'): bacteria transformation, plasmid extraction, plasmid analysis
- Plant transformation
- Long read sequencing (ONT) (doi: 10.1017/qpb.2021.18 and 10.3390/genes11030274 and 10.1186/s12864-026-12623-z)
- Pore-C (chromatin structure analysis) (doi: 10.17504/protocols.io.rm7vz9mmrgx1/v1)
- RNA extraction for RNA-seq
- Extraction of metabolites
- Various assays to analyze flavonoids
- In vitro plant cultivation
Protocols for various methods are shared through our GitHub repository.
Bioinformatics Methods
- De novo genome assembly (doi: 10.1186/s13104-023-06459-z and 10.1371/journal.pone.0216233 and 10.20944/preprints202508.1176.v2)
- Genome sequence annotation (doi: 10.1371/journal.pone.0294342 and 10.1186/s12864-022-08452-5 and 10.1186/s12864-026-12623-z)
- Read mapping and variant calling (doi: 10.3390/plants9040439)
- RNA-Seq analysis (doi: 10.1186/s12864-018-5360-z)
- Co-expression analyses (doi: 10.1186/s12864-025-12061-3)
- Synteny analysis (doi: 10.1371/journal.pone.0280155 and 10.1111/nph.19341 and 10.1101/2025.10.06.680802)
- Functional annotation of genes (doi: 10.3390/plants9091103 and 10.1371/journal.pone.0294342 and 10.1186/s12864-022-08452-5)
- Tool development (doi: 10.1186/s12864-022-08452-5 and 10.1186/s12864-023-09877-2 and 10.1101/2025.10.10.681656)

Ongoing and completed genome sequencing projects
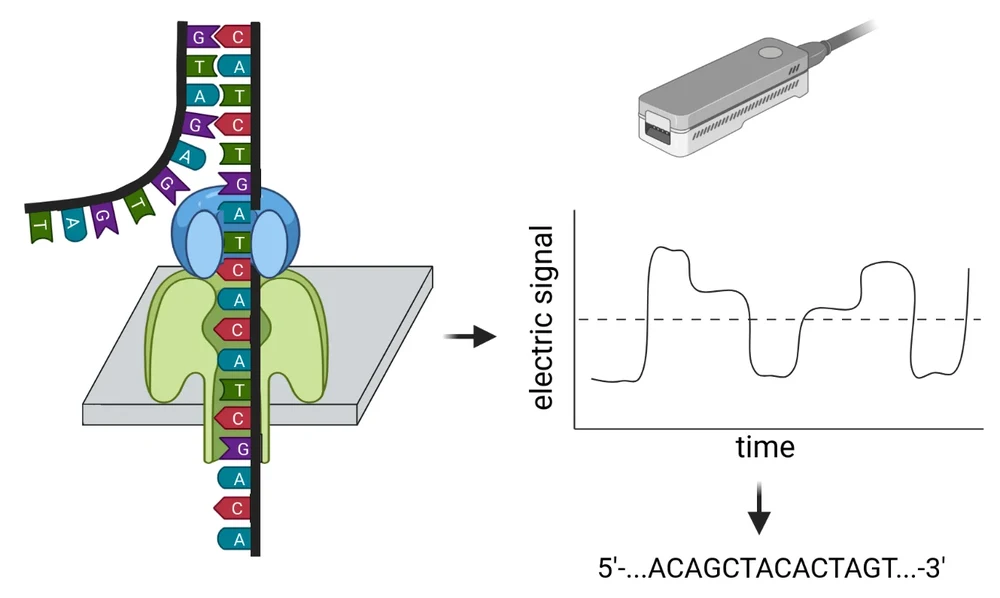
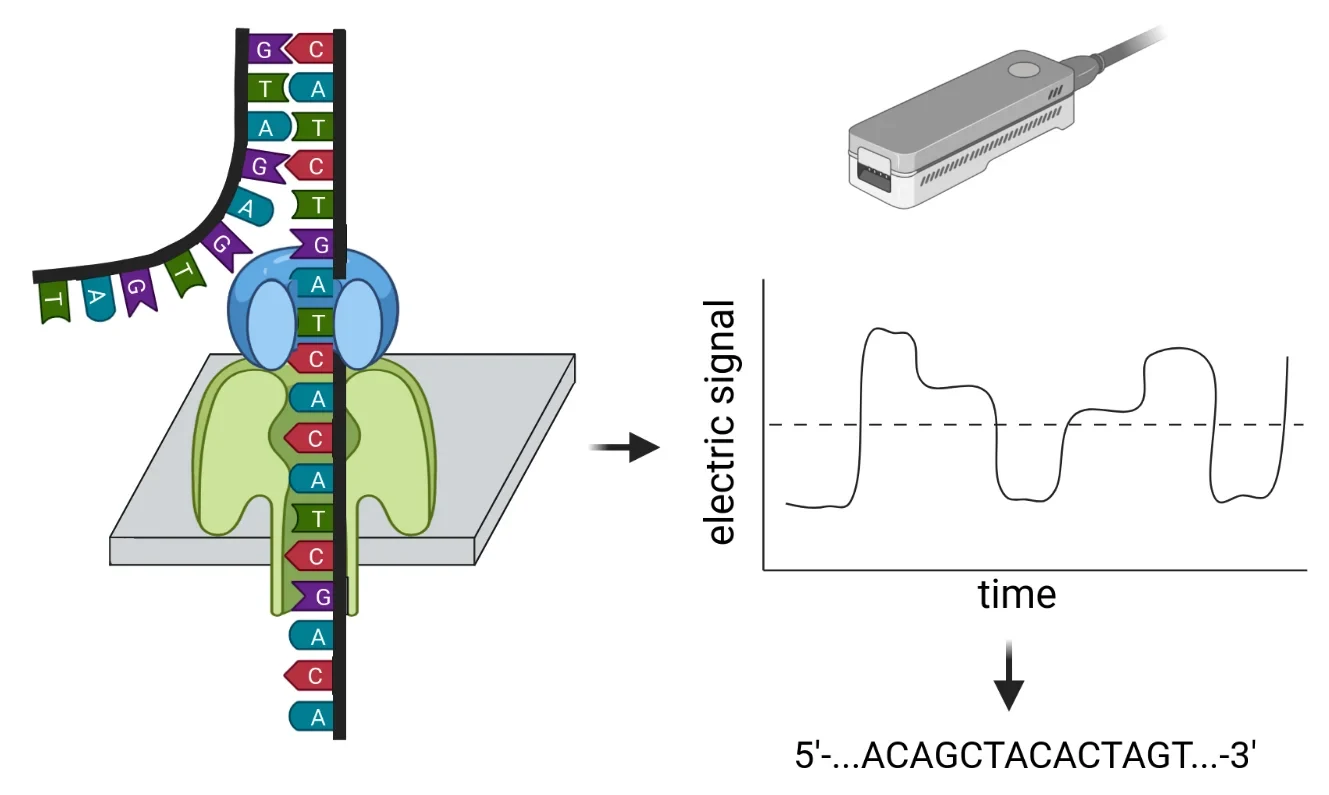
This website provides an overview of our group's current and completed genome sequencing projects. We utilize the Nanopore sequencing technology from Oxford Nanopore Technologies (ONT). In this process, a single DNA molecule (or RNA molecule) is pulled through a protein embedded in a synthetic membrane. While a single-stranded DNA passes through this protein, the bases of the DNA selectively block the movement of small charged particles (ions) through the protein. This generates an electrical signal that can be measured. Later, this signal is translated into a DNA sequence because each combination of DNA bases produces a distinct electrical signal. Further details about this sequencing technology and its capabilities were recently described in a review article (Pucker et al., 2022). For sequencing, we use MinIONs, which are small, portable sequencers. Students have the opportunity to participate in genome sequencing projects as part of various courses, enabling them to gain hands-on experience with this sequencing technology.
The completion of these genome sequencing projects and subsequent publication of the genome sequence and all related data unfortunately often takes several months. As a principle, we publish all data in appropriate repositories (e.g., ENA) and also make all methods freely available (e.g., scripts via GitHub). If you are interested in our ongoing projects, please feel free to contact us via email. In most cases, access to the genome sequence should be possible before its official publication.
In general, we are interested in collaborations for sequencing additional plant genomes. However, our research is heavily constrained by the Nagoya Protocol. Therefore, we must limit our activities to species that are native to Germany and can be collected here. Species from abroad would need to have been present in Germany before 2014 (e.g., in botanical gardens), and we require documents that clearly confirm this. Some source countries provide their genetic resources internationally for unrestricted use (e.g., Germany), allowing us to work with plants from these countries. Unfortunately, we must decline projects involving plants that are subject to the Nagoya Protocol.
Ceratophyllum submersum
Ceratophyllum submersum (soft hornwort) is an aquatic plant that is very common in (eutrophic) ponds in Germany. We analyzed the plastome (subgenome of the chloroplast) of this species to determine its relationship to other plant species (Meckoni et al., 2023).
Digitalis purpurea
Digitalis purpurea (floxglove) is known as a medicinal plant, but also appears frequently as horticultural plant in gardens due to its colorfull and large flowers. We are interested in the flavonoid biosynthesis of this plant species with a particular focus on anthocyanins which are responsible for the redish pigmentation and also the name of the species. Read about the details in our Preprint on bioRxiv (Horz et al., 2025).
Tropaeolum majus
Tropaeolum majus is well known for the high content of medicinal compounds. We sequenced the genome and screened the annotated gene set for players of the flavonoid biosynthesis (Friedhoff et al., 2024).
Victoria cruziana
Victoria cruziana (giant water lily) is the biggest attraction in the Botanical Garden in Braunschweig. The plant is famous for huge floating leaves that can carry the weight of a small child. It produces beautiful flowers that open at night. We are interested to understand the flavonoid biosynthesis in this aquatic plant species. A first publication about the genome sequence and the flower color chnage of this species is available on bioRxiv (Nowak, Harder, Meckoni et al., 2025). More details about our V. cruziana research are available here.
Urtica dioica
Urtica dioica (stinging nettle) was the medicinal plant of the year in 2022. It is well known for a high flavonoid content and often used to produce tea. We sequenced the genome and screened the annotated gene set for players of the flavonoid biosynthesis (Wolff et al., 2026).
Aquilegia vulgaris
Aquilegia vulgaris (common columbine) is a horticultural plant that can be seen in many gardens showing a variety of flower colors. We are interested to understand the molecular basis of different flower colors (Friedhoff et al., 2024).
Rubus armeniacus
Rubus armeniacus (blackberry) is a fruit producing plant species that is common throughout Germany. It is well known for the black and sweet fruits that it produces. We are interested to study the formation of anthocyanins, a subgroup of the flavonoids, in blackberry. We sequenced the genome to access the gene set encoding players of the flavonoid biosynthesis (Wolff et al., 2026).
Theobroma cacao
Theobroma cacao is a well known crop species producing crucial ingredients for the chocolate production. The genome sequence provides access to genes determining these properties (Marin Recinos et al., 2024).
Centaurea cyanus
Cornflower is well known for the striking blue flower color. The genome sequence provides insights into the involved genes (Dassow et al., 2025).
Begonia manicata
This is a popular horticultural plant displaying structures with bright anthocyanin pigmentation.The genome sequence provides insights into the genes responsible for pigmentation of red structures on top of leaves (Fischer et al., 2026).
Tacca chantrieri
This plant is famous for showy flowers with almost black pigmentation. The genome sequences enables new research into a particular enzyme involved in anthocyanin biosynthesis (de Oliveira & Pucker, 2026).
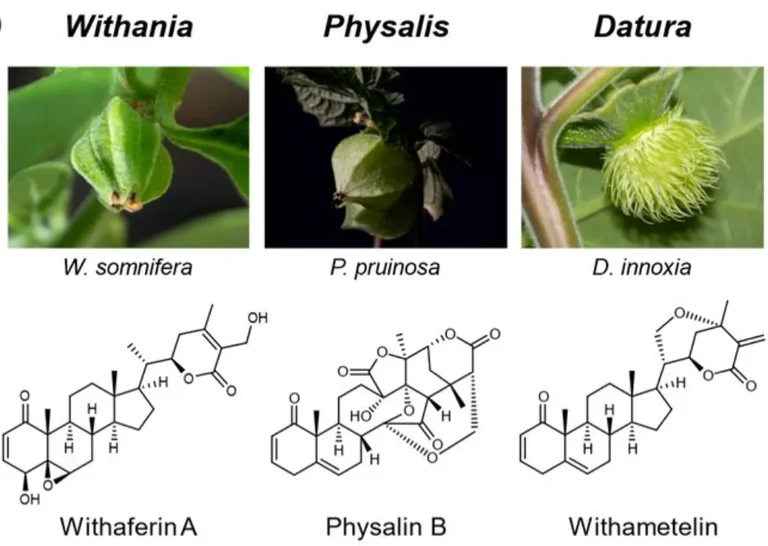
Phylogenomic elucidation of withanolide biosynthesis in Nightshade plants
Withanolides are specialised metabolites from Nightshade plants with potent antiproliferative activity, which possess a long history of human usage as traditional medicine. More than 700 natural withanolides have been isolated so far, and many representatives are biologically active. Little is known, however, how these structurally diverse steroidal lactones are synthesised in plants. In preliminary work, we identified a putative withanolide biosynthetic gene cluster by analysis of all publicly available genome and transcriptome data of withanolide producers. In this project, building on a collaboration between a plant biochemistry group and a bioinformatics group, we want to explore this hypothesis and leverage it for the phylogenomic elucidation of withanolide biosynthesis. Specifically, we will verify our already identified gene candidates by heterologous expression and virus-induced gene silencing. In addition, to further understand the evolution of different withanolide subclasses, we will sequence three additional genomes of the withanolide producers Withania somnifera, Datura innoxia and Nicandra physalodes. From this sequence data, we will select and evaluate gene candidates based on synteny, phylogenetic relationship, and co-expression that might be responsible for the generation of structural subclasses. Based on the sequence data and the biosynthetic genes gained by this approach, we will then be able to reconstruct the evolutionary history of withanolides and their different subclasses. Thus, our project will provide fundamental insights into the molecular basis and evolution of withanolide biosynthesis. On a long term, these findings will help to better harness the medicinal properties of this structurally varied natural product class for drug development.
We are conducting this project with funding from the DFG in coopration with Jakob Franke's reserach group. First results have been published (Hakim et al., 2025).
FlavonoidFriday
On Fridays, our group shares recent publications and fascinating details about the flavonoid biosynthesis via Twitter and LinkedIn under the hashtag "#FlavonoidFriday". A collection of all posts is available in the FlavonoidFriday GitHub repository. Due to their contribution to flowers and other plant parts, flavonoids are widely distributed and visually striking.
Plants produce a plethora of specialized metabolites to cope with numerous environmental challenges. Examples are terpenoids, flavonoids, and betalains. We are particularly interested in flavonoids which are responsible for the pigmentation of many blue or red flowers. Flavonoids can be separated into several subgroups including flavonols, anthocyanins, proanthocyanidins, and flavones. There are multiple reasons why working on the flavonoid biosynthesis is promising.
1) The core of the flavonoid biosynthesis is a well understood model system for the regulation of biosynthesis pathways in plants. Decades of work revealed insights into the different enzymes, but also into the transcriptional regulation of the involved genes. Even Nobel Prizes have been won in association with the flavonoid biosynthesis.
2) Flavonoids have enormous potential in biotechnological applications as natural colorants and due to their high nutritional value. While the general biosynthesis pathway is well conserved across distantly related plant species, their are species-specific differences in the modification of flavonoids. Detailed knowledge about the flavonoid biosynthesis in (orphan) crop species can pave the way to a healthy nutrition. Genome editing or breeding methods can be used to improve crops with respect to their nutritional value.
3) Visible phenotypes resulting from the knock-out or increased activation of flavonoid biosynthesis genes can be helpful in the identification of promising mutants. Visible phenotypes were one reason why the flavonoid biosynthesis was established as a model system. However, coloration is not only important for basic research. Plants with altered expression of certain flavonoid biosynthesis genes show often fascinating colorations and are favored in horticultural plant species.
Generally, we are interested in the discovery of promising biosynthesis pathways for biotechnological applications. This is not restricted to the flavonoid biosynthesis, but instead using this model system to develop new genome mining approaches. Different methods for the identification biosynthetic pathways are combined including screens and comparisons of plant genome sequences.
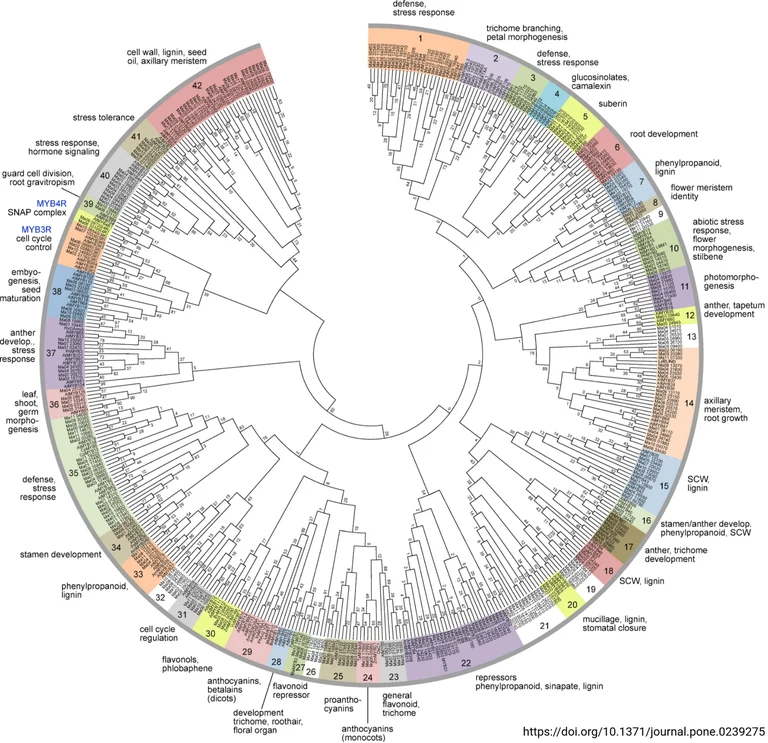
MybMonday
On Mondays, our group shares recent publications and fascinating details about the MYB transcription factors via Twitter and LinkedIn under the hashtag "#MybMonday". A collection of all posts is available in the MybMonday GitHub repository.
Data Publications
Please find a list of our publicly available datasets below:
- Genome sequence and annotation of Victoria cruziana. doi: 10.60507/FK2/5DS0JZ.
- Supplementary data to Evolutionary Dynamics of the Proanthocyanidin Biosynthesis Gene LAR. doi: 10.60507/FK2/UKAAO0.
- Supplementary data supporting the study: "Out of the blue: Family-wide loss of anthocyanin biosynthesis in Cucurbitaceae". doi: 10.60507/FK2/ZNVSLA.
- Digitalis purpurea genome sequence and annotation V2. doi: 10.60507/FK2/4KUKXI.
- Structural and functional annotation of Physalis grisea and Physalis pruinosa. doi: 10.24355/dbbs.084-202409130931-0.
- Genome sequence and annotation of Withania somnifera v2. doi: 10.24355/dbbs.084-202503200640-0.
- Gene expression data sets of selected plant species. doi: 10.24355/dbbs.084-202306231402-0.
- Gene Expression Analysis Across Plantae. doi: 10.24355/dbbs.084-202501230512-0.
- Collection of plant gene expression data. doi: 10.24355/dbbs.084-202409160820-0.
- Genome sequence and annotation of the medicinal plant Tropaeolum majus. doi: 10.24355/dbbs.084-202410230959-0.
- Haplotype-resolved genome sequence of Cassava (Manihot esculenta) cultivar COL40. doi: 10.24355/dbbs.084-202409230712-0.
- Tacca chantrieri genome sequence and annotation. doi: https://doi.org/10.60507/FK2/6SOMER.
- Purple and White Aquilegia vulgaris genome sequence and annotation V2. doi: https://doi.org/10.60507/FK2/GKLMOG.
- Begonia manicata genome sequence and annotation. doi: 10.60507/FK2/MOR7FZ.
- Rubus armeniacus genome sequence and annotation. doi: 10.60507/FK2/L7LG2L.
- Genome sequence and annotation of Urtica dioica. doi: 10.60507/FK2/1XHSZ3.
- (more data publications are coming soon)
PuckerLab Plasmid Sequencing
Do you regularly produce plasmids that need to be checked by sequencing? We might be able to help with our full plasmid squencing based on ONT long-reads. In order to participate, you need to reach out to Boas Pucker to get your research group registered. Registration is currently possible for research groups at the University of Bonn and collaborators. Registration of your research group is important to ensure that your samples are labeled with the correct ID.
There are some limitations:
- We can only analyze plasmids if you have an expected sequence.
- It is not possible to analyze different clones of the same transformation experiment simultaneously, because it would not be possible to identify which clone is the correct one if not all are correct. Different inserts in the same backbone are not a problem though.
- Sequencing only takes place once we have collected sufficient plasmids (>20).
There are two components of the sample submission:
- Plasmid: Please submit your plasmids with a concentration of 50 ng/µL in TE buffer (or water) in a 1.5mL reaction tube. The tube should be labeled with a four digit/character code, which starts with the identifier of your research group followed by three digits/characters. Only numbers 0-9 and upper case characters A-Z are permitted. (The regular expression would look like this: [RESEARCH-GROUP-ID] [0-9A-Z]{3} ). We recommend to use continuously increasing numbers for samples from your research group to allow alpha-numerical sorting of your samples. Example for a research group with A as their group identifier: A001, A002, A003, A004, A005, ...
- FASTA: Since our data analysis is based on comparing the reads against your expected sequence, we need a FASTA file with the expected sequence of your plasmid. Please name the file according to the ID written on your tube (e.g. "A0001.fasta"). You will get access to a Sciebo folder to drop off your FASTA files prior to the sequencing run. Without this FASTA file, processing of your sample is not possible.
Results will be shared via Sciebo on the day after the sequencing run. Please download your files and ensure you have proper backups, because we are going to delete the data after some time. You can expect a number of different result files including a read mapping against your expected sequence. This read mapping can be visualized in IGV (https://igv.org/app/) to check all positions in detail. We are going to release more detailed instructions soon. If you have questions or suggestions for improvements, please reach out. We would love to learn about your experience!
While our plasmid sequencing is free of charge for you, we would appreciate if you would cite the underlying workflow in your resulting publications:
de Oliveira J.A.V.S., Ng V., Wolff K., Pucker B. (2026). NanoPlasmiQC: Full plasmid sequencing with ONT long-reads and automatic data analysis. bioRxiv 2026.04.01.715842; doi: 10.64898/2026.04.01.715842.